Sleep, Melatonin, DHEA, AIDS and Sudden Infant Death Syndrome
Copyright ã 1995 Revised from
1985 by James Michael Howard.
Support
of this theory:
"DHEAS concentrations increased after melatonin therapy. Moreover, a
tendency towards a higher DHEAS/cortisol ratio was found after 6 months of
treatment."
Neuroendocrinology Letters 2002 Apr; 23 Suppl 1: 17-9
Some Direct
Support, missed in the past:
“DHEA administration induced a significant (P < 0.05)
increase in rapid eye movement (REM) sleep, whereas all other sleep variables
remained unchanged compared with the placebo condition.” “Spectral analysis of five selected EEG bands
revealed significantly (P < 0.05) enhanced EEG activity in the sigma
frequency range during REM sleep in the first 2-h sleep period after DHEA
administration.”
American Journal of Physiology, Endocrinology and Metabolism 1995; 31: E107-E113
(This is from my
explanation of sleep of 1985: “The deeper levels of sleep would be produced by
higher concentrations of melatonin. The REM sleep would be produced by DHEA as
it reciprocates with melatonin. REM
sleep would increase as SWS sleep decreases toward morning.” (“A Theory of the Control of the Ontogeny and
Phylogeny of Homo sapiens by the
interaction of Dehydroepiandrosterone and the Amygdala,” Copyright 1985, James
Michael Howard)
My explanation of sleep is derived from my
foundation hypothesis that the adrenal hormone, DHEA (dehydroepiandrosterone),
is necessary for transcription and duplication of DNA in all tissues, and that
the pineal gland hormone, melatonin, acts in concert with DHEA. In my
copyrighted document of 1985, I developed a sleep mechanism, dependent on an
interaction between melatonin and DHEA. Since 1985, I have expanded it with
further support, however, the original mechanism
remains unchanged and viable.
My theory states that the brain is most
sensitive to DHEA and melatonin. This is due to enhanced development of
"chemical doorways" (receptors) for entrance of DHEA into brain
tissues. These receptors may be stimulated by melatonin. This is how I account
for increased transcription and metabolic activity in the brain compared to
other tissues.
"Brain is characterized by high
metabolic activity and exhibits two to three times the transcriptional activity
of other tissues." (J. Neurochemistry 1991; 56: 812)
Sleep Mechanism
Very Brief Abstract of Theory: In daytime
concentrations, DHEA, when acting with specific proteins, is the molecule of
consciousness, i.e., activation of the nervous system. During deep sleep,
concentrations of DHEA are reduced; only enough DHEA is produced to maintain
autonomic functions. Melatonin is the molecule of sleep. Melatonin causes
sleep, because it reduces production of DHEA. During sleep, reduced nighttime
levels of DHEA reciprocate with melatonin to produce either slow-wave sleep
(SWS) or rapid eye movement (REM) sleep. These low productions of DHEA are
higher during REM sleep, and lower during SWS. As sleep occurs, melatonin is
"used up," and DHEA increases. During consciousness, DHEA is used;
melatonin increases, but is not released till DHEA levels decline to a low
level prior to sleep.
The following diagram of sleep stages is
based on classic sleep charts. The undulating line represents the transition
into sleep from consciousness; the firtst slow wave sleep (SWS), followed by
the first REM sleep, etc. The first SWS is the
deepest, that is, stage four; subsequent SWS is less deep as sleep progresses.
REM sleep approaches concisousness and is often associated with short periods
of awakenings. As sleep time progresses, REM increases until full awakening
occurs.
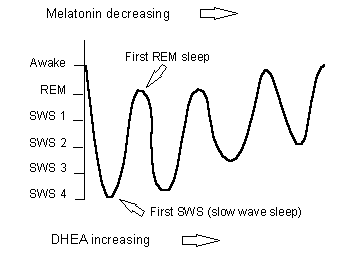
The
following quotations demonstrate the connection of melatonin with sleep:
"We presently regard the normal pattern of secretion [of melatonin] in
healthy subjects as one that shows a 24-hr cycle of marked circadian
rhythmicity, with low circulating levels in serum during the day and rises to
maximal concentrations during the night." (Biological
Psychiatry 1988; 23: 406). A lot of journal citations, since
this one, support the idea that melatonin induces sleep including this one
which tested melatonin’s ability to induce sleep. "These data indicate
that orally administered melatonin can be a highly potent hypnotic agent; they
also suggest that the physiological increase in serum melatonin levels, which
occurs around 2100 h daily, may constitute a signal initiating normal sleep
onset." (Proc. Natl. Acad. Sci. USA 1994; 91:
1824). I did not make the original connection of melatonin and sleep; it
is my mechanism that explains how melatonin induces sleep, and that DHEA is the
other side of the mechanism.

When
DHEA naturally begins to decline around age twenty-five, all activities in all
tissues should begin to decline. DHEA is reduced during sleep. Therefore, brain
activity, as measured by electroencephalogram (EEG), should be reduced during
sleep and even more in aged animals. The following quotation supports my
hypotheses. (Don’t confuse slow wave sleep with EEG slow waves. In this quotation,
EEG slow waves represent reduced brain activity. I attribute this reduced brain
activity to reduced DHEA; it is even slower during sleep in aged animals.)
"In the awake
stage, EEGs in the aged rats consisted predominantly of slow waves when compared
with those in the young rats. ...The relative powers of the cortical and
hippocampal waves in the aged rats differed from those of the young rats during
the slow-wave sleep stage. These findings suggest that the slowing of the EEG
in the aged rats during the awake stage may be related to decreased
brain activity associated with aging, and that the irregular burst waves in the
cortical EEG in the aged rats appear to correlate with the changes observed in
age-related human sleep patterns." (Physiology and Behavior 1988; 44:
389)
I suggest melatonin reduces DHEA at night.
This reduction in DHEA is achieved by inhibitory actions of melatonin on
production of the hormone, prolactin. Prolactin (PRL) secretion may be shown to
specifically stimulate production of DHEA. That is, when melatonin release is
high, prolactin is reduced, which reduces DHEA production. Melatonin binding
sites (chemical doorways or receptors) develop early in gestation (pregnancy)
in the hypothalamus of the developing brain. The hypothalamus is the site of
prolactin production. Melatonin receptors also develop early in the
suprachiasmatic nucleus; I will explain a little later that the suprachiasmatic
nuclei (a specific area in the brain) is directly
involved in stopping melatonin release.
"The results suggest that
melatonin has a single effect in alteration of gestation in mink; i.e. the
prevention of prolactin secretion." (J. Reproduction and Fertility
1990; 89: 423)
"Melatonin treatment appeared
more efficient than an artificial photoperiod in reducing plasma prolactin
concentrations." (J. Endocrinology 1986; 108: 287)
"In the present study melatonin
binding sites were first found on the 15th day of gestation, at which time
specific binding was limited to the pituitary. ...Moreover, melatonin binding
sites are identifiable over the suprachiasmatic nuclei from day 18 of gestation
[in the fetus]." (Endocrinology 1991; 128: 2083)
Another hormone, also produced in the same
area as prolactin, can stimulate DHEA production. It is called
adrenocorticotropic hormone (ACTH). (These hormones are both made in the hypothalamus, migrate to the pituitary gland, where they are
released into the blood stream to be carried to the adrenal glands.) In some
text books, ACTH is given credit for being the only stimulator of DHEA and
another adrenal hormone, cortisol. At this point, the important connection is
the relationship of melatonin and DHEA with prolactin.
"Dehydroepiandrosterone
formation was increased 3.5 fold and five [fold] by adrenocorticotropic hormone
and prolactin respectively." (Am. J. Obstetrics and Gynecology
1987; 156: 1275)
"These findings indicate that
the majority of serum androgens in young baboons is of
adrenal origin. Therefore, we conclude that PRL [prolactin], in addition to
ACTH, may also be an adrenocorticophic factor in baboon infants. However in
contrast to ACTH, the action of PRL on the adrenal is apparently specific for
androgen [DHEA] production." (Endocrinology 1985; 117: 1968)
Sleep Mechanism and Temperature
When melatonin is in greatest concentration,
I suggest slow wave sleep (SWS) occurs. Therefore, during stage 4 (the deepest
slow wave sleep), melatonin should be in greatest
concentration and DHEA least. The next paragraph and citations demonstrate the
connection of DHEA with metabolism. Melatonin and DHEA cycle throughout the
night, with melatonin declining and DHEA increasing. During times when DHEA
rises, but does not initiate consciousness, REM sleep occurs. That is, this is
a time when some brain activity is stimulated by DHEA, but is not sufficient to
cause awakening. DHEA stimulates metabolism, therefore, temperature should fall
during slow wave sleep.
"This exaggerated temperature fall is accompanied by a
pronounced increase in Stage 4 slow wave sleep." (Psychophysiology
1987; 24: 200)
The basis of this rests in the findings that
DHEA directly affects resting metabolism. This is the result of the fact that
DHEA stimulates mitochondrial respiration. (Mitochondria are subcellular
"organoids" that are the site of energy production in cells.) During
SWS, DHEA should be at its lowest, hence, metabolic processes should be at
their lowest level. (In the second quotation below, you can see that DHEA
affects protein synthesis. This is an indication that DHEA affects
transcription; transcription has to occur in order for protein synthesis to
occur.)
"There is growing clinical and
experimental evidence that dehydroepiandrosterone... plays an important
regulatory role in intermediary metabolism by inhibiting the storage of dietary
energy as fat." (J. Nutrition 1987; 117: 1287)
"These findings indicate that
mitochondrial respiration is the earliest factor affected by DHEA and may be
associated with protein synthesis." (J. Nutrition 1991; 121:
240)
Since oxygen (O2) consumption is a direct
measurement of mitochondrial respiration, oxygen consumption should be highest
in REM sleep and increase as DHEA increases during the night. That is, SWS
decreases as sleep progresses through the night; DHEA rises
as SWS decreases. Therefore, oxygen consumption, during sleep, should be
highest during REM sleep and increase during the night.
"In both feeding modalities O2
consumption was always highest in REM sleep, intermediate in stage 2 and lowest
in SWS." (Physiology & Behavior 1991; 49: 1159)
Evidence suggests that sleep patterns are
related to mental retardation. My theory states that mental retardation results
from reduced DHEA for brain growth. I suggest the reason for the dip in DHEA of
early and mid-childhood, in the chart above, results from the large amount of
melatonin of the same period. That is, melatonin and DHEA act together for
brain growth at this time. Mental retardation may also be due to reduced
melatonin at this time. However, the levels of DHEA may be deduced from levels
of REM sleep. The following findings support my theory that DHEA is involved in
REM sleep, i.e., retarded individuals exhibit less REM
sleep.
"In mentally retarded subjects,
patterns of REM sleep ratios and REM densities below normal have been observed
on several occasions. ...Already in mentally retarded infants, developmental
slowness can be observed which is reflected in the REM sleep patterns of
neonates. On the other hand, gifted children have a higher REM sleep rate, REM
density and R [an index of cerebral ability to organize information]. ...In
premature babies, a sensorial stimulation schedule applied during waking
accelerates the appearance of sleep patterns similar to those of infants born
at normal term age, and especially leads to an increase in REM density during
activated sleep." (Physiology & Behavior 1990; 47: 1273)
It is common knowledge in Departments of
Pediatrics and Children’s Hospitals that "failure to thrive" infants
respond readily to holding. That is, frequent holding and attention to these
babies brings about dramatic changes in many of them. I suggest the handling
and attention stimulate prolactin and, therefore, DHEA production in these
infants. This is the same sort of phenomenon mentioned in the quotation above
(sensorial stimulation).
I suggest melatonin (MLT) induces sleep
because it may be our natural narcotic. That is, MLT loosely satisfies one set
of criteria of the classic narcotic drug (agonist). The reason MLT does not
directly contain the structures of a powerful narcotic is due to evolution.
Animals that evolved using more powerful sleep inducers were more prone to
predation; their genes simply did not survive.
"In all potent narcotics, it is
possible to recognize three molecular characteristics that are believed to be
related to the mode of actions of the drugs: (1) a tertiary basic nitrogen, (2)
a quaternary carbon separated from the basic nitrogen by an ethylene chain
(C-C-C=C), and (3) a phenolic hydroxyl or a ketone." (Encyclopædia
Britannica 1984; 12: 842)
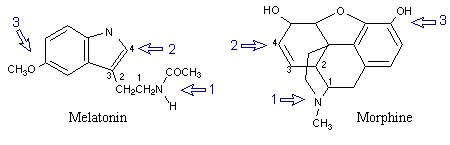
I have
demonstrated that melatonin suppresses prolactin release. Hence, MLT reduces
DHEA production, because PRL stimulates DHEA. I think PRL rebounds to
suppression by MLT. Therefore, a short "time lag" exists in the
prolactin rebound to MLT suppression. This is seen in the following diagram
(this is a generalization in that these smooth lines really consist of many
peaks and troughs in production.) The PRL rebound to MLT periodically induces
DHEA production; this produces a cycling of DHEA with MLT during sleep. (The
periodic peaks in DHEA during this time are usually insufficient to stimulate
consciousness, however, brain activation occurs and this is called REM or dream
sleep.) The following diagram is taken partly from The New England J. of
Medicine 1987; 316: 1550; after diagram on page 1551. I have added
another diagram to this that shows DHEA during one day (Best and Taylor’s
Physiological Basis of Medical Practice, 11th. ed., Williams and Wilkins,
1985; after Figure 8.33, page 886).
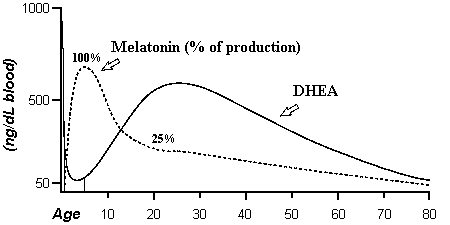
My
hypotheses regarding the relationship of prolactin to DHEA during sleep suggest
prolactin and DHEA should follow a specific pattern. Since sleep is a dangerous
time, that is, sufficient DHEA must be available to maintain the brain stem, prolactin release must be triggered soon after the
initial reduction of prolactin by melatonin (SWS). To avoid awakening during
the nocturnal releases of prolactin that stimulate DHEA, prolactin secretion
must decline during the time of REM. That is, prolactin must be produced soon
after SWS and stop production during REM. This pattern has been found in
healthy, young humans. Prolactin rebounds to melatonin suppression.
"Sleep onset was rapidly
followed by an increase in secretion [of prolactin], and awakenings coincided
with an immediate offset of active secretion [of prolactin]. Analyzing the
association between secretory pulses and sleep stages demonstrated that PRL
secretory rate is low at the time of rapid eye movement sleep onset." (Sleep
1994; 17: 20)
Cortisol and DHEA production are usually
synchronous. Therefore, changes in levels of cortisol may also indicate changes
in DHEA production. Remember that prolactin stimulates DHEA production and
compare the following quotation with the one just above. The last sentence in
this quotation is remarkable, because it may verify that DHEA, not cortisol, is
causing awakening.
"The pattern of nocturnal
cortisol secretion appeared to be synchronized with the periodicity of sleep:
rapid eye movement (REM) sleep was found to be primarily present when cortisol
concentrations were decreasing, indicating a diminished or absent secretory
activity of the adrenals at that time; wakefulness and Stage 1 sleep, by
contrast, were associated with increasing plasma cortisol concentrations.
Furthermore, the enhanced adrenal secretory activity usually preceded the
occurrence of light sleep or wakefulness, which is in accord with a wakening
effect of plasma cortisol." (Biological Psychiatry 1986; 21:
1415)
(It is part of my theory that narcotic
addiction results from the same mechanism just described. That is, narcotic
addiction results from an increase in DHEA production, triggered by the drugs.
This increased DHEA would increase stimulation in the brain and in the body;
early in addiction, the extra DHEA enhances the pleasure centers that the drugs
activate. Eventually, increased drug use increases DHEA. I suggest that at some
point narcotics would then be used primarily to ward off the effects of DHEA
over-stimulation. I explain in my article on AIDS that DHEA over-stimulation
occurs at viral infection; this is the cause of viral symptoms like those that
come with influenza infection. We don’t like this feeling caused by our natural
over-production of DHEA; neither do people addicted to narcotics.)
Narcotics are known to stimulate endorphin
production; hence, I suggest melatonin stimulates endorphin production. Since
MLT is produced primarily at night, an inverse relationship should exist
between endorphin and prolactin production at night. MLT should stimulate
endorphins, which would then be followed by PRL; this is the time lag rebound.
This may be seen in the system examined in the following quotation. A time
relationship of endorphin and prolactin is demonstrated that supports my
hypotheses. (Melatonin production would be higher during times of short days,
i.e., winter.)
"Based on these results, we
conclude that endogenous opioid peptides [endorphines] are essential in
initiating and maintaining nocturnal PRL surges in pregnant rats." (Endocrinology
1991; 129: 925)
"The levels of endorphin were
highest under short days (15-fold change), and inversely related to the changes
in the plasma levels of prolactin (120-fold change)." (J. Endocrinology
1990; 127: 461)
"A daily rhythm of endorphin
content within both the AHA [anterior hypothalamus] and MBH [medial basal
hypothalamus] of animals exposed to short photoperiod coincided with this
prolonged nighttime rise in pineal melatonin content, although a causal relationship
between the two was not established." (Endocrinology 1985; 117:
141)
During daytime, melatonin would be released
in very small amounts, if at all, while DHEA is high and vice versa at night.
(Daytime release of MLT may cause what we call depression; other hormones such
as testosterone can interfere with DHEA availability. It may be testosterone
that causes the long-term type of depression called "dysphoria.")
Since MLT would still be synthesized in the pineal gland during the day, a
large amount would be available in a large release at the first sleep of the
night; hence the first sleep is the deepest slow wave sleep. Release of MLT is
inhibited during the day, because of a nerve stimulation produced by the
superior cervical ganglia of the sympathetic nervous system to the pineal gland
(Endocrine Physiology 1985; C.R. Martin, Oxford University Press, New
York, page 866). The superior cervical ganglia (a group of nerves in a specific
spot) receive an input, from the eyes, that keeps it active in inhibiting
release of MLT from the pineal. This input also comes from the suprachiasmatic
nuclei; remember the melatonin receptors on this structure? If MLT binds to
this, it would help close down the superior cervical ganglia. This is why banks
of very bright lights, or less light from "full spectrum bulbs," help
people with SAD (seasonal affective disorder). Melatonin production increases
in winter because of the reduced sunlight. That is, these people produce MLT
inappropriately during the day, which reduces their DHEA and makes them
fatigued and depressed. What you should also know is that normal people
maintain the sleep-consciousness cycle without light input from the sun or
artificial light. This normal cycle is due to DHEA. That is, the DHEA which
normally rises, after MLT-PRL have gone full cycle,
stimulates parts of the brain which stimulate the superior cervical ganglia.
Hence, while light will augment the DHEA-MLT circadian cycle, it is normal
levels of DHEA and MLT that maintain the cycle. To my knowledge, micronized,
wax coated DHEA during the day has not been used to help people with SAD, but I
think it might be beneficial to them.
In 1990, it was reported that receptors for
delta-nine-tetrahydrocannabinol (THC) had been located in the brains of several
species, including humans (Proc. Natl. Acad. Sci. USA 1990; 87:
1932). THC is the psychoactive chemical in marijuana or cannabis. I think THC
receptors are really DHEA receptors. The following diagram shows the basic
structural similarity which may allow THC to use DHEA receptors:
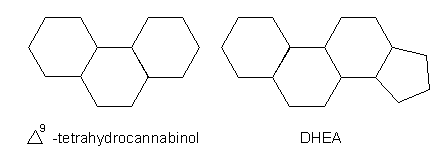
"Generally high densities in forebrain and cerebellum
implicate roles for cannabinoids in cognition and movement. Sparse densities in
lower brainstem areas controlling cardiovascular and respiratory functions may
explain why high doses of delta-nine-tetrahydrocannabinol are not lethal."
(reference in paragraph above)
Upon reading the location of THC receptors, I
immediately knew I had further support for my sleep mechanism. Note in the
quotation above that binding densities of THC are "sparse" in
"the lower brainstem areas controlling cardiovascular and respiratory
functions." Since I think all brain activity depends on DHEA, the small
densities of these receptors in the brainstem fits, that is, cardio-respiratory
functions are so important they evolved to function on small amounts of DHEA to
remain active. This developed out of the sleep mechanism, when DHEA is very
low. Animals that evolved the ability to maintain brainstem functions on low
amounts of DHEA during sleep, survived. (When infants do not produce sufficient
DHEA to maintain brainstem stimulation, they succumb to sudden infant death
syndrome.) The increased levels of these receptors in the upper brain may be
the reason that brains evolved into larger and more complex structures over
time; increasing receptors increase the growth promoting input of DHEA.
Acquired Immune
Deficiency Syndrome and Sleep Mechanism
The next part of this treatise will refer
mainly to the journal article: AIDS 1995; 9: 1043-1050;
quotations below are from this, unless I cite another journal. This is an
article about sleep in HIV infection and includes an interesting introduction.
(I will include sufficient amounts of my explanation of AIDS during this essay
to connect this article with my sleep mechanism; if you want further detail
about AIDS, see my explanation of AIDS at Anthropogeny.com. I will use CD4 to
mean CD4+ lymphocytes.) The finding that "THC" receptors are
"generally high in forebrain and cerebellum" suggests to me that DHEA
is very important for "cognition and movement." In a proven case
where DHEA is low and continues to decline, such as HIV infection progressing
to AIDS, cognitive abilities are reduced proportionately. That is, as DHEA
declines, cognitive ability declines because of insufficient DHEA for the
receptors; it is like parts of a complex machine not getting enough power to
run some parts. When the parts with increased receptors are shut down, activity
may increase in lower brain structures that depend on lower levels of DHEA. For
example, in advanced AIDS the activity of the front part of the brain is
reduced while an area called the "basal ganglia" increase in activity
(Science 1988; 239: 587). This same phenomenon occurs in
schizophrenia (Am. J. Psychiatry 1985; 142: 564), and DHEA is
significantly low in schizophrenia (Biological Psychiatry 1973; 6:
23).
The AIDS journal article states:
"The differences between our two HIV-positive groups were not
statistically significant with regard to their cognitive-motor performances.
They both performed significantly worse when compared with controls; even
though these two groups differ in CD4 counts. However, those with CD4 counts
[greater than] >400 X 106/l tended toward performing slightly better than
those with CD4 counts [less than] <400 X 106/l." Cognitive and motor
abilities should decline as DHEA declines. DHEA has declined in both groups;
DHEA is reduced in HIV+ and lower in AIDS (JAMA 1989; 261: 1149).
In my article on evolution and AIDS, I explain that all tissues compete for
DHEA. Of importance here is that the brain and immune system are competing for
DHEA. My article on AIDS contains citations showing that DHEA rises upon HIV
infection and another that shows that DHEA causes CD4 cells to decline. I
suggest that upon HIV infection, this DHEA rise and subsequent CD4 decline is
the natural reaction to viral infection (look this up in my AIDS article for
details). Since the group, with CD4 counts >400, has better cognitive and
motor abilities than those with <400, I interpret this to mean that the
group with >400 is using more DHEA for brain function and less for immune
function. There is less of a DHEA response to viral infection in the group with
>400 cells. There is less DHEA in the group with CD4 >440 for the immune
system; they are using it for brain function. Below, you will read that this
use of DHEA for brain function, instead of for immune function, will cause
problems in another area, that is, sleep disturbance.
This report compared various aspects of sleep
in 13 HIV negative men (controls) and 24 HIV positive men. Findings about sleep
included: "When sleep parameter data were analyzed by HIV status, REM
latency emerged as the only sleep variable significantly different between
HIV-seropositive subjects and HIV-seronegative controls." (REM latency is
the time it takes to reach the first REM sleep (see my
diagram above); this follows the first SWS.) REM latency is 72.6 min in
controls, 87.7 min in CD4 <400, and 103.2 in CD4 >400. The first slow
wave sleep, I suggest, comes from the first release of melatonin. Remember that
melatonin, by reducing DHEA, causes a DHEA rebound to maintain sufficient DHEA
for the brainstem. DHEA is highest in controls,
therefore, the rebound is fastest. Again, for clarity, DHEA is used to fight
the HIV and it reduces CD4s, therefore, those with less CD4 are showing a
greater response of DHEA for their immune system. That means that those with
less CD4s are producing more DHEA in response. Hence, the group with <400
CD4s have more DHEA for the melatonin response at night than those with
>400. However, the group with <400 still have a longer REM latency than
controls because they have less of a DHEA response to the first SWS. Those with
CD4s >400 are responding with less DHEA because their brains have used up a
lot of their DHEA for cognitive and motor abilities. Therefore, it takes longer
for them to respond to melatonin at night, and they have the longest REM
latency.
Further in the article: "Analyses
revealed that subjects with CD4+ counts <400 had sleep parameters similar to
controls. However, subjects with CD4+ counts >400 demonstrated a greater
amount of SWS in the middle third of the night and final third of the night
when compared with controls. In addition, these subjects had significantly
fewer awakenings during the night." also ..."The foregoing analyses
in combination with inspection of the data indicates that HIV infection exerts
a major effect on the amount and distribution of NREM [nonREM; same as SWS]
sleep stages 3 and 4. ...Of note is the overall tendency for HIV-positive
subjects to have increased SWS relative to controls, especially in the final
third of the night." Since both seropositive groups have reduced DHEA,
they both should have increased SWS sleep. Since the group, with CD4 >400,
has less DHEA, according to my work, they should have less awakening and
increased SWS throughout the night. The similarity of sleep between those with
CD4 <400 and controls is probably due to the fact that controls and those
with lower CD4s probably recruit DHEA in a similar manner. That is, a control
exposed to the HIV should produce a large increase in DHEA; those with CD4s
<400, I suggest, have done just that. You should know that many individuals
who succumb to AIDS, produce this response, that is, low CD4s, but they cannot
maintain DHEA levels sufficiently long enough to remove the virus (see my
article on AIDS ). Individuals who progress to AIDS,
do not produce enough of the reserve form of DHEA called DHEA sulfate. DHEA
comes from DHEAS (citation in my article on AIDS ).
Sudden Infant Death Syndrome and Sleep Mechanism
SIDS occurs during sleep. I think SIDS
results from inadequate DHEA during sleep. In my short remarks about mental
retardation, I explained that the dip in DHEA of childhood, along with the rise
in melatonin, occur for the purpose of brain growth. The brain uses so much
that measurable levels of DHEA decline; the body is also using the same
mechanism for growth at this time in the competition for DHEA. The J. of
Endocrinology 1991; 128: 8 reports the following: "(1) After birth and until the third month of neonatal life, low
levels of MLT are secreted by the pineal gland without any pronounced diurnal
rhythm. (2) From about the third month of life, there is a pronounced diurnal
variation in MLT secretion by the pineal and circulating MLT levels are high, particularly
at night. (3) After the first year or two of life, as children age, there is a
profound reduction in the circulating levels of MLT, particularly at
night." The following quotation will demonstrate that the peak incidence
of SIDS occurs just prior to, but continues through, this period of increased
production of MLT. Additionally, the next quotation will show that this is a
time of the lowest measurable levels of DHEA. MLT is high; this increases the
receptors for DHEA, DHEA is absorbed for use and its levels in the blood are
reduced.
"...it is now accepted that the
peak incidence [of SIDS] occurs at about 10 weeks of age and that it is
uncommon at less than 3 weeks and greater than 9 months." (JAMA
1989; 262: 1565)
"During the first day of life
the concentration of plasma DHA [DHEA] (unconjugated) is high, averaging 920
ng/100 ml in both sexes. During the first month of life, concentrations fall
reaching a mean of approximately 70 ng/100 ml by age 1 year." (Adrenal
Androgens, A.R. Genazzani, et al., Raven Press, 1980, page 30)
Infants are using DHEA for brain and body
growth and development at this time. Herein lies the
problem for SIDS victims. These infants are using their DHEA at a much greater
pace than normal infants, because they are growing much faster than normals in
both brain and body. Therefore, their background DHEA at night must be very
low, to the point that their brainstem cannot function. I think this is part of
the explanation of SIDS.
"Somatic [body] growth and
brain weight were significantly greater in SIDS than controls." (J.
Neuropathology and Experimental Neurology 1991; 50: 29)
I have suggested that when DHEA is being
used, the measurable levels decline; hence the decline in DHEA of childhood.
However, the adrenal glands of SIDS victims appear normal (Int. J. Legal
Medicine 1994; 106: 244). It follows that the significant increase
in body growth and brain weight in SIDS victims should also be reflected in
reduced melatonin levels. It has been determined that melatonin is
significantly reduced in SIDS.
"A significant correlation was
observed for melatonin levels in different body fluids from the same
individual. After adjusting for age differences, CSF melatonin levels were
significantly lower among the SIDS infants than among those dying of other
causes." (Forensic Sci. Int. 1990; 45: 171)
The findings reported in the following
quotation are that SIDS victims are "intrauterine growth retarded."
"SIDS infants weighed, on
average, 85 g less at birth than their siblings and 164 g less compared with
babies in nonaffected sibships. When birth weights were standardized for
gestational age, most of the weight difference between SIDS infants and
siblings was due to a shorter gestational age of SIDS infants, while the
difference between surviving siblings of SIDS infants and births from
nonaffected sibships remained. All births in sibships with a SIDS infant were
intrauterine growth retarded. " (American J.
Epidemiology 1995; 142: 84)
I suggest SIDS results from the following
mechanism. Upon birth, these "intrauterine growth retarded" infants
experience a rebound of growth which literally uses up their melatonin and
DHEA. When DHEA is too low during sleep to maintain activity in the cardio-respiratory
mechanism of their brainstems, they expire. In old age, DHEA and melatonin both
decline to very low levels. Perhaps, when a specific cause cannot be identified
in death during sleep this should be called "sudden elderly death
syndrome."